Cronache
Coronavirus. Al 100% è mutato. Ma ora si può capire come si diffonderà e dove
Studio dell’Università di Cambridge e dalla famosa rivista scientifica PNAS spiegano come sia mutato radicalmente. Tarro: la strada è la terapia antivirale
Un passo importante è stato realizzato per capire come il Coronavirus ha attecchito nel corpo umano e quali strategie siano utili per debellarlo. Quali altre no.
Uno studio riportato dalla prestigiosa Università di Cambridge e realizzato da studiosi dell’università britannica oltre che da ricercatori inglesi e tedeschi ha tracciato le mutazioni genetiche del Coronavirus, così come si è diffuso in Cina e dall'Asia all'Australia, fino all'Europa e al Nord America.
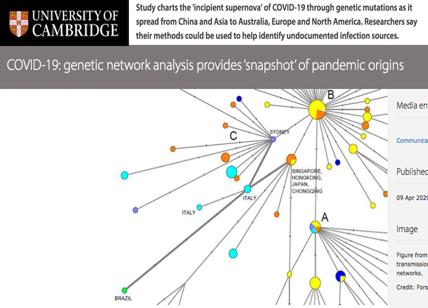
I ricercatori di Cambridge, usando le reti genetiche, hanno ricostruito i primi "percorsi evolutivi" del virus negli esseri umani e come l'infezione si è diffusa da Wuhan all'Europa e al Nord America.
“Ci sono troppe mutazioni rapide per rintracciare ordinatamente un albero genealogico COVID-19”, ha affermato uno degli autori dello studio, il genetista Peter Forster, “abbiamo utilizzato un algoritmo di rete matematica per visualizzare contemporaneamente tutti gli alberi plausibili".
“Queste tecniche sono note soprattutto per la mappatura dei movimenti delle popolazioni umane preistoriche attraverso il DNA. Pensiamo che questa sia una delle prime volte in cui sono stati utilizzate per tracciare le vie di infezione di un Coronavirus come il COVID-19”.
La ricerca ha rivelato tre distinte “varianti” di COVID-19, costituite da gruppi di lignaggi strettamente correlati, che sono state etichettate come variante “A”, “B” e “C”. I dati dei genomi dei virus campionati da tutto il mondo sono quelli circolati tra il 24 dicembre 2019 e il 4 marzo 2020.
Forster e colleghi hanno scoperto che il COVID-19 scoperto nei pipistrelli, di tipo “A”, il cosiddetto “genoma virale umano originale” era presente a Wuhan, ma sorprendentemente non era il tipo di virus predominante della città. Sono state trovate mutazioni della versione “A” in pazienti provenienti dagli Stati Uniti e dall'Australia.
Il principale tipo di virus di Wuhan sarebbe invece la versione “B”, prevalente nei pazienti di tutta l'Asia orientale. Tuttavia, la variante non ha viaggiato molto oltre la regione e non ha ottenuto ulteriori mutazioni.
La variante europea è invece quella che viene denominato come variante “C”, presente nei primi pazienti in Francia, Italia, Svezia e Inghilterra. La variante “C” è assente nel campione cinese dello studio, ma è stata trovata a Singapore, Hong Kong e Corea del Sud.
La variante “A”, più strettamente correlata al virus trovato nei pipistrelli e nel pangolino, è descritta dai ricercatori come “la radice dell'epidemia”. Il tipo “B” deriva dal tipo “A”, separato da due mutazioni, quindi la variante “C” è a sua volta una “figlia” della variante “B”. Il virus di tipo B di Wuhan potrebbe essersi adattato immunologicamente o ecologicamente a una vasta parte della popolazione dell'Asia orientale. Il virus a quel punto potrebbe essere mutato per superare la resistenza al di fuori dell'Asia orientale. Si assiste a un tasso di mutazione più lento di questa variante in Asia orientale che altrove, almeno questo è quanto sembra sia avvenuto inizialmente.
Gli studiosi tornano sulla prima immissioni del virus in Italia. Si sarebbe procurata tramite la prima infezione tedesca documentata il 27 gennaio. Un'altra prima via di infezione italiana è collegata a un “cluster di Singapore”. E’ ovviamente una delle prime immissioni documentate in Italia, più difficile se non impossibile dimostrare o sapere se ce ne sono state altre in precedenza.
La tecnica utilizzata e la rete genetica che è stata tracciata, sostengono i ricercatori, potrebbe essere importante per capire le vie di infezione, le mutazioni e i lignaggi virali in modo da aiutare a prevedere come e dove si diffonderà la malattia, capire cioè i futuri punti caldi globali della trasmissione e delle sue impennate. Una strategia fondamentare per prevedere le mosse del virus e non solo inseguirlo.
I ricercatori Peter Forster, Lucy Forster, Colin Renfrew e Michael Forster che firmano l’analisi hanno prodotto un approfondimento su 160 genomi virali, ottenuti da pazienti umani, utilizzando anche un software di classificazione che traccia oltre 1.000 genomi. Lo studio pubblicato l’8 aprile su una delle riviste scientifiche più note a livello internazionale, PNAS (Proceedings of the National Academy of Sciences of the United States of America), evidenzia come il metodo di indagine potrebbe essere utile per identificare anche le fonti di infezione non documentate.
Dato lo scenario tracciato dai ricercatori, acquisisce un valore importante anche l’analisi fatta di recente al professor Giulio Tarro, virologo di fama internazionale, allievo e collaboratore di Albert Sabin, il padre del vaccino contro la poliomelite. Tarro, 2 volte candidato al Nobel e insignito nel 2018 dall'Associazione internazionale dei migliori professionisti del mondo (IAOTP) del premio Usa di virologo dell'anno, come raccontato in Italia da Affaritaliani, non è uno studioso amato dai virologi dei salotti nostrani.
Ma il consiglio di Tarro è di indirizzarsi verso una terapia antivirale efficace e di non pensare prevalentemente al vaccino, visto che il virus ha, come sembra, una variante cinese e una europea o padana. Tarro: “Sarà complicato avere un vaccino che funzioni in entrambi i casi esattamente come avviene per i vaccini antinfluenzali che non coprono tutto”.



